Portal hypertension is an abnormal increase in pressure within the portal venous system. It is most commonly caused by cirrhosis, where fibrosis and regenerative nodules increase resistance to blood flow through the liver.
Understanding portal hypertension is essential because many major complications of chronic liver disease — oesophageal varices, splenomegaly, thrombocytopenia, ascites and hepatic encephalopathy — are directly or indirectly related to increased portal pressure.
This article is the foundation of the hepatology cluster. Read it alongside Child-Pugh Score Explained and MELD Score Explained to build a complete picture of chronic liver disease.
Portal hypertension is the haemodynamic basis of many complications of cirrhosis.
Learning Objectives
- Define portal hypertension
- Describe normal portal venous circulation
- Explain how cirrhosis causes portal hypertension
- Understand portosystemic collateral formation
- Explain why varices form and why they are dangerous
- Explain why splenomegaly and thrombocytopenia occur
- Understand the basic mechanism of ascites in portal hypertension
- Recognise the key clinical consequences of portal hypertension
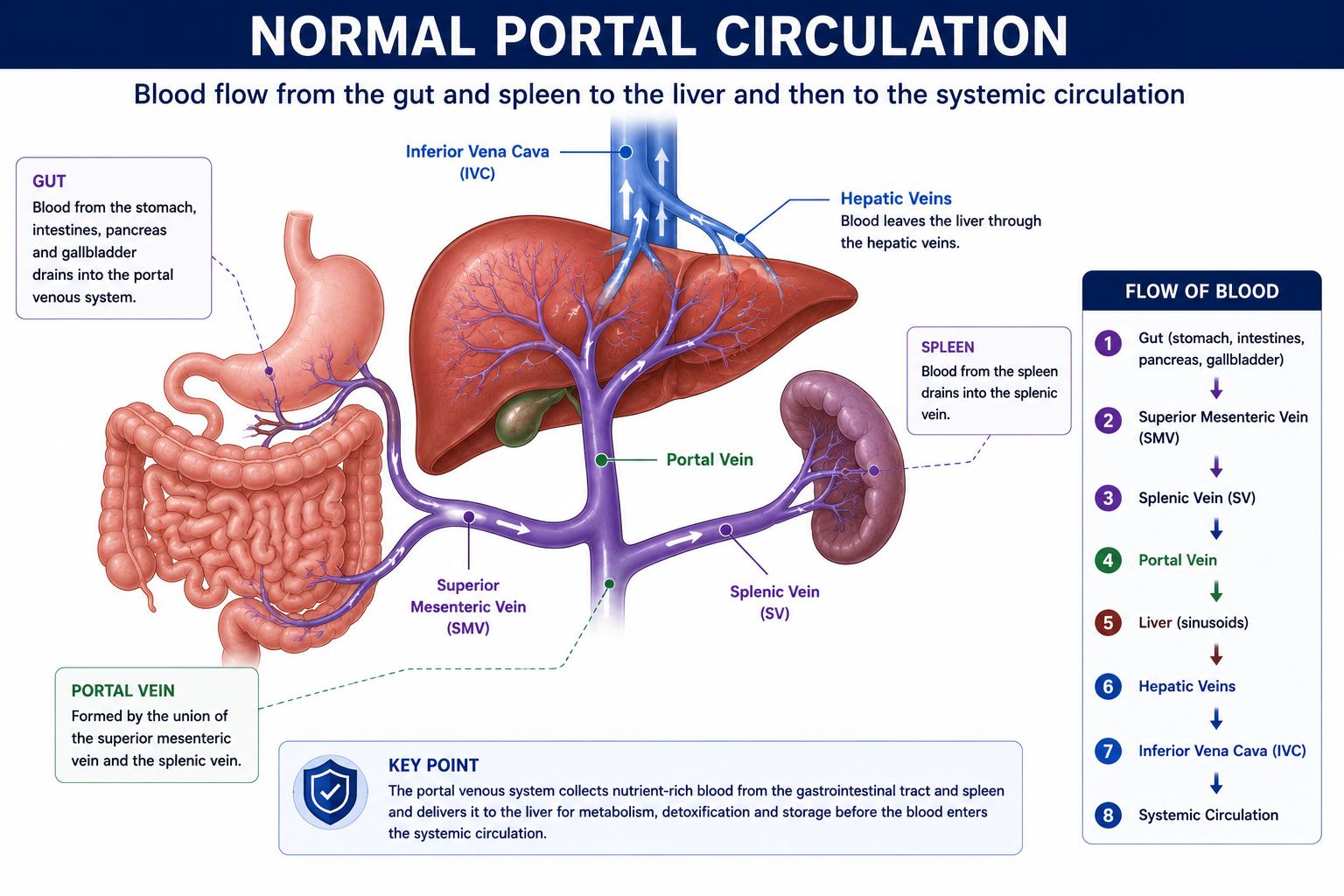
What Is the Portal Venous System?
The portal venous system drains blood from the gastrointestinal tract, spleen and pancreas into the liver before it enters the systemic circulation.
The portal vein is formed mainly by the union of two vessels:
- Superior mesenteric vein — draining the small intestine and proximal colon
- Splenic vein — draining the spleen and pancreas
This arrangement allows nutrients, toxins and gut-derived substances — including bacteria and their products — to pass through the liver for processing before entering the systemic circulation. The liver is therefore the first organ to receive and handle gut-absorbed material.
What Is Portal Hypertension?
Portal hypertension means increased pressure within the portal venous system. It is defined by the hepatic venous pressure gradient (HVPG) — the difference between portal venous pressure and hepatic venous pressure.
| HVPG | Meaning |
|---|---|
| 1–5 mmHg | Normal |
| >5 mmHg | Portal hypertension |
| ≥10 mmHg | Clinically significant portal hypertension — varices begin to develop |
| ≥12 mmHg | Increased risk of variceal bleeding |
The two clinically important HVPG thresholds are ≥10 mmHg (clinically significant portal hypertension — where complications begin to develop) and ≥12 mmHg (where the risk of variceal bleeding increases significantly). Both thresholds are frequently tested in examinations.
Why Is Portal Hypertension Dangerous?
Portal hypertension becomes clinically important because elevated portal pressure forces blood to seek alternative routes, bypassing the liver and creating complications that can be life-threatening. Many of the most serious consequences of advanced cirrhosis are directly or indirectly related to raised portal pressure — making it a central concept for medical students studying liver disease.
The most important consequences are:
- Variceal bleeding — from dilated oesophageal and gastric collaterals; can be catastrophic
- Ascites — fluid accumulation in the peritoneal cavity
- Hepatic encephalopathy — neurotoxins bypassing the liver via portosystemic shunting
- Splenomegaly — from splenic venous congestion
- Hypersplenism and thrombocytopenia — from platelet sequestration in the enlarged spleen
Portal hypertension is the haemodynamic link between cirrhosis and many of its major complications. Understanding portal hypertension is therefore the essential first step before studying varices, ascites, or hepatic encephalopathy in depth.
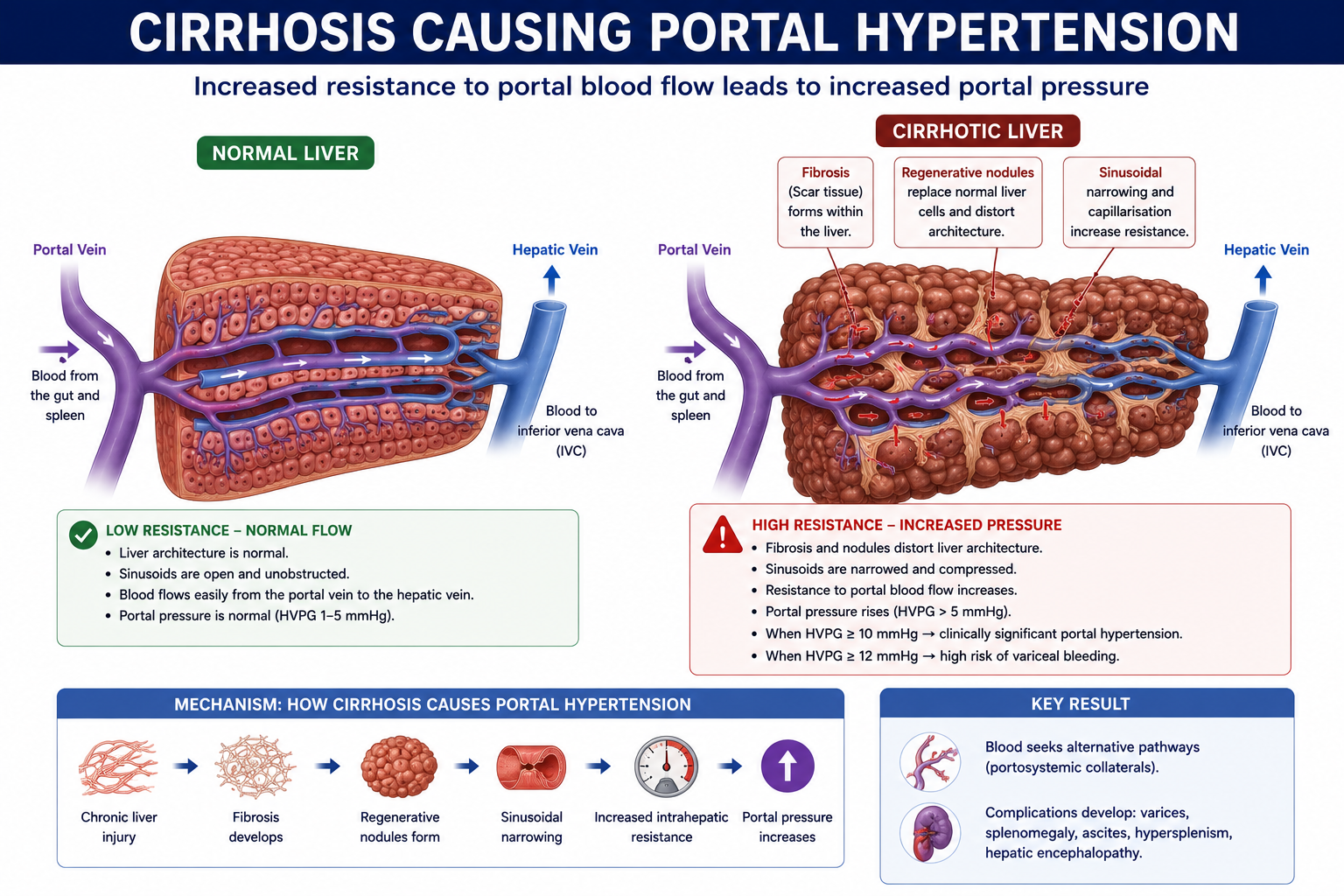
How Does Cirrhosis Cause Portal Hypertension?
Cirrhosis causes portal hypertension through two distinct but related mechanisms that both increase resistance to portal blood flow.
1. Structural Resistance
Hepatic fibrosis, regenerative nodules and architectural distortion narrow the sinusoidal and vascular channels within the liver. The normal sponge-like sinusoidal architecture is replaced by a fibrotic scaffold that physically impedes blood flow. This is the dominant and largely irreversible component of resistance in established cirrhosis.
2. Dynamic Resistance
Endothelial dysfunction within the cirrhotic liver leads to reduced nitric oxide (NO) production. Nitric oxide normally promotes sinusoidal vasodilation. When NO is deficient, hepatic stellate cells and vascular smooth muscle remain tonically contracted — causing sinusoidal vasoconstriction that further increases intrahepatic resistance. Unlike structural resistance, this component is potentially reversible.
Structural resistance (fibrosis, nodules) is largely irreversible once established. Dynamic resistance (sinusoidal vasoconstriction from reduced nitric oxide) is potentially modifiable — this is the basis for the use of non-selective beta-blockers (e.g. propranolol, carvedilol) and other pharmacological agents to reduce portal pressure.
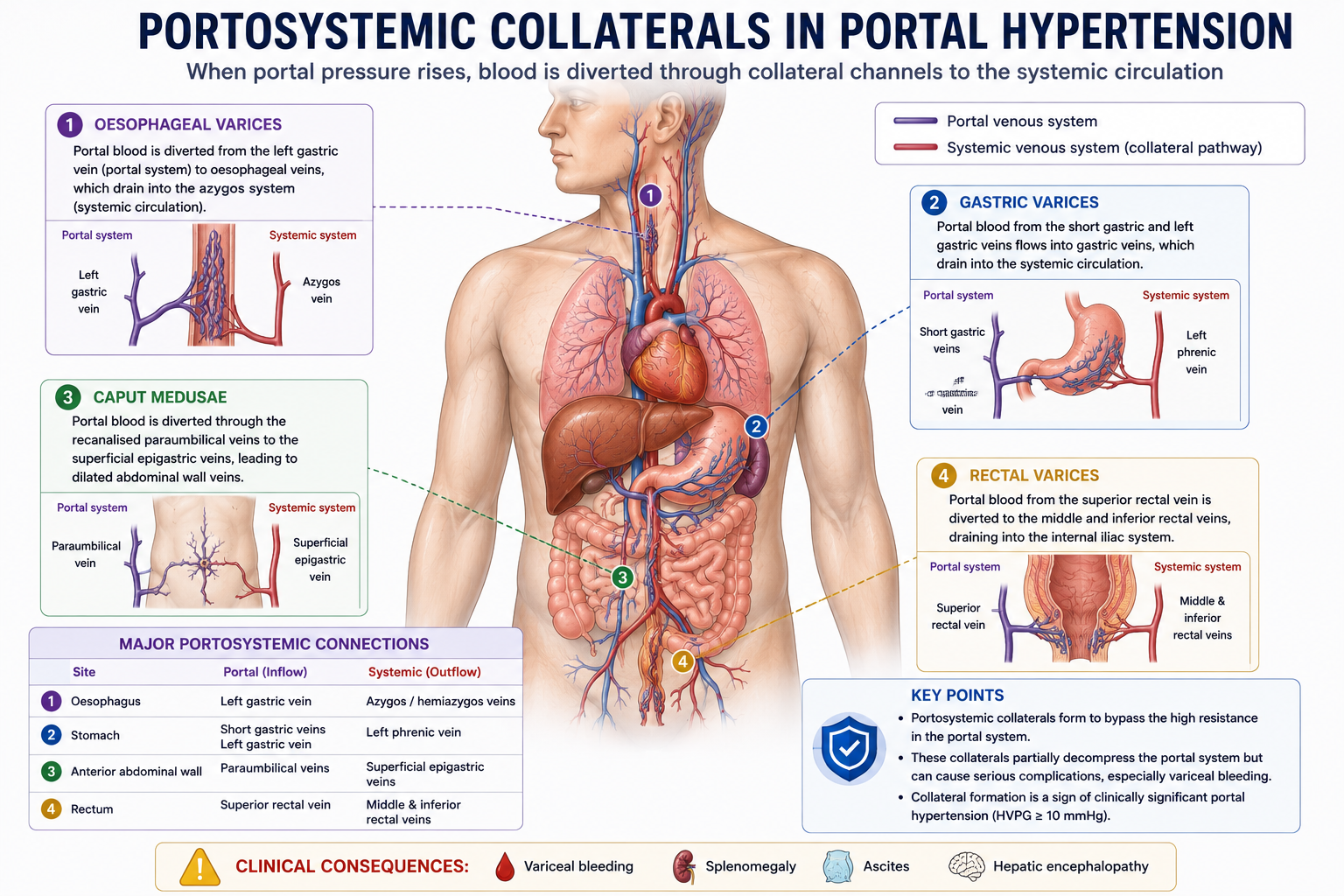
Portosystemic Collaterals
When portal pressure rises, portal blood seeks alternative pathways back to the systemic circulation. These bypass channels are called portosystemic collaterals. They develop at sites where portal and systemic venous beds normally communicate.
| Site | Clinical Manifestation |
|---|---|
| Gastro-oesophageal junction | Oesophageal and gastric varices |
| Umbilicus | Caput medusae (recanalised paraumbilical veins) |
| Rectum | Rectal varices (not haemorrhoids) |
| Retroperitoneum | Retroperitoneal collaterals |
Portosystemic collaterals partially reduce portal pressure by diverting blood away from the liver. However, they create serious complications — especially variceal bleeding at the gastro-oesophageal junction.
Collateral vessels allow portal blood to bypass the liver entirely. While this partially decompresses the portal system, it also allows ammonia and other gut-derived toxins to enter the systemic circulation without normal hepatic detoxification. This portosystemic shunting is the primary mechanism by which portal hypertension contributes to hepatic encephalopathy.
Varices
Varices are dilated collateral veins that develop when portal blood is diverted into systemic venous channels under high pressure. They are most clinically important at the gastro-oesophageal junction, where oesophageal and gastric varices carry the highest risk of catastrophic bleeding.
Why Varices Are Dangerous
- Thin venous walls — unlike arteries, veins lack a thick muscular media and are prone to rupture under sustained pressure
- High intraluminal pressure — portal blood is transmitted at elevated pressure into the collateral channels
- Poor support from surrounding tissue — submucosal varices at the gastro-oesophageal junction have minimal external support
- Risk of massive bleeding — variceal haemorrhage carries significant mortality even with modern endoscopic and pharmacological management
Varices typically develop when HVPG is ≥10 mmHg.
Variceal bleeding risk increases significantly when HVPG is ≥12 mmHg.
Not all varices bleed — risk also depends on variceal size, red wale signs on endoscopy, and severity of liver disease.
Splenomegaly and Hypersplenism
Portal hypertension increases pressure throughout the portal venous system, including the splenic vein. The sequence of events from raised portal pressure to thrombocytopenia is direct and important to understand:
This is why a low platelet count is often one of the earliest and most accessible laboratory clues to portal hypertension in a patient with chronic liver disease — sometimes appearing before overt varices or ascites are detectable.
Why Do Platelets Fall in Cirrhosis?
Thrombocytopenia in cirrhosis is multifactorial. Several mechanisms contribute simultaneously:
- 1Splenic sequestration — the enlarged spleen traps and destroys platelets at an accelerated rate. This is the dominant mechanism in most patients with portal hypertension.
- 2Reduced thrombopoietin (TPO) production — thrombopoietin is the primary stimulus for platelet production and is synthesised mainly by the liver. A diseased liver produces less TPO, reducing platelet production from the bone marrow.
- 3Bone marrow suppression — particularly in alcohol-related liver disease, where alcohol directly suppresses megakaryocyte development. Chronic illness and nutritional deficiencies also contribute.
- 4Immune-mediated platelet destruction — in some liver diseases (e.g. autoimmune hepatitis, viral hepatitis), antiplatelet antibodies may contribute to platelet destruction.
Low platelets in cirrhosis are not simply due to bone marrow failure. Portal hypertension and hypersplenism are major contributors. This is clinically important — treating portal hypertension (e.g. with TIPS or transjugular intrahepatic portosystemic shunt) can significantly improve platelet counts by reducing splenic sequestration.
Ascites
Ascites is the accumulation of fluid within the peritoneal cavity. Portal hypertension is the central haemodynamic driver. The mechanism involves a cascade of neurohormonal events:
The development of ascites marks a critical transition — from compensated to decompensated cirrhosis — and is associated with a significant worsening of prognosis. Hypoalbuminaemia (reducing oncotic pressure) and increased lymphatic production further contribute alongside the neurohormonal mechanisms.
The full mechanism of ascites formation — including the role of SAAG, ascitic fluid analysis, and management — is covered in detail in Ascites Explained.
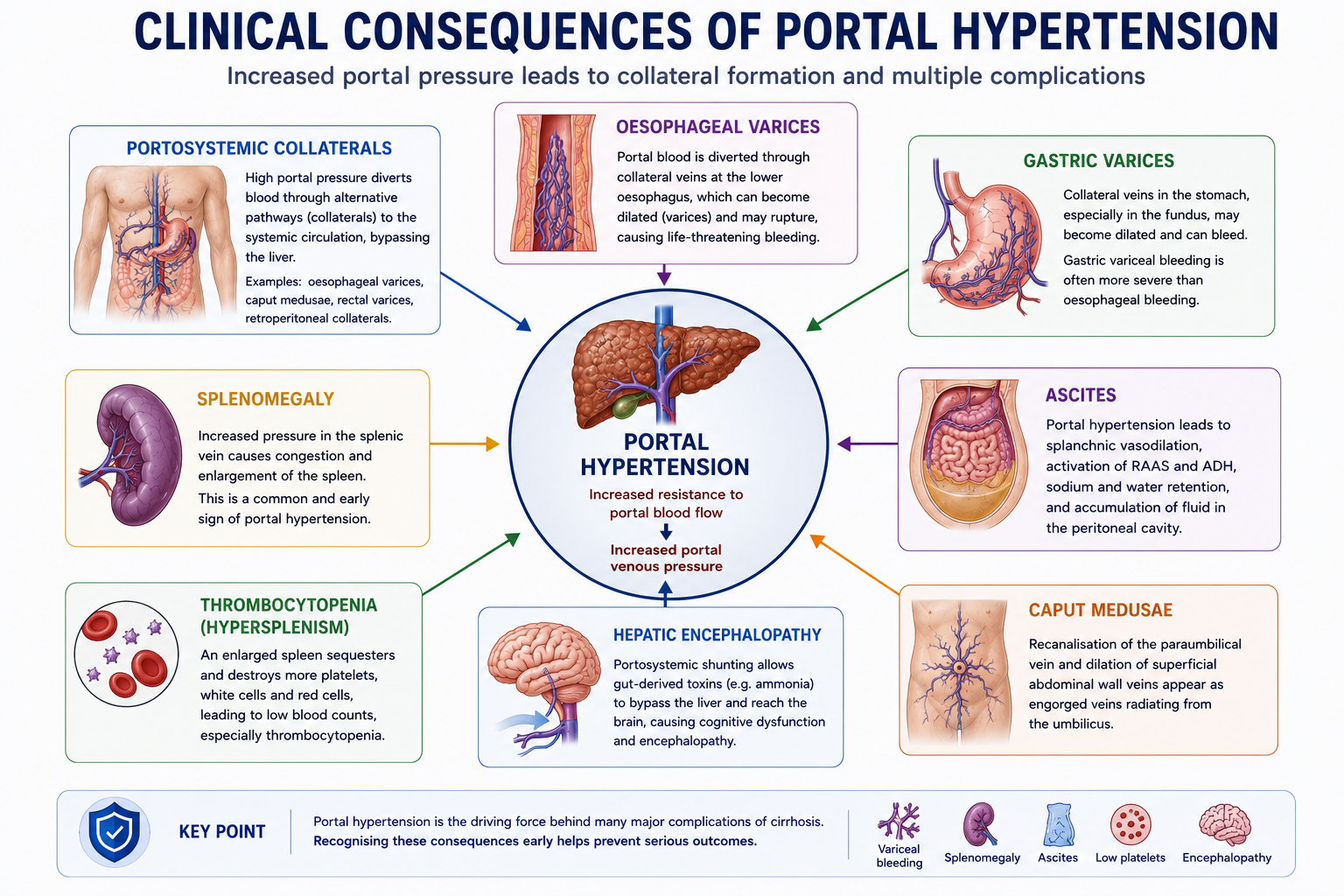
Clinical Consequences of Portal Hypertension
Portal hypertension produces a broad range of clinical consequences, each with a distinct underlying mechanism.
| Consequence | Mechanism |
|---|---|
| Oesophageal varices | Portosystemic collateral formation at the gastro-oesophageal junction |
| Gastric varices | Collateral formation through gastric veins |
| Splenomegaly | Splenic venous congestion from raised portal pressure |
| Thrombocytopenia | Hypersplenism and reduced thrombopoietin production |
| Ascites | Portal hypertension, splanchnic vasodilation and renal sodium retention |
| Caput medusae | Recanalised paraumbilical veins carrying portal blood to the abdominal wall |
| Hepatic encephalopathy | Portosystemic shunting of gut-derived toxins bypassing hepatic detoxification |
Hepatic Encephalopathy and Portosystemic Shunting
Hepatic encephalopathy occurs when gut-derived neurotoxins — especially ammonia — bypass hepatic detoxification and enter the systemic circulation, where they reach the brain and cause neurological dysfunction.
In portal hypertension, portosystemic collaterals divert portal blood around the liver. The gut constantly produces ammonia through bacterial breakdown of nitrogenous compounds. Normally the liver converts ammonia to urea for excretion. When portal blood bypasses the liver via collaterals, this ammonia detoxification step is lost.
Clinical features may include sleep disturbance, personality change, confusion, altered behaviour, reduced level of consciousness, and in severe cases, coma.
Portosystemic shunting → ammonia bypasses liver → systemic ammonia rises → brain toxicity → hepatic encephalopathy.
This is why controlling portal hypertension (e.g. with beta-blockers or TIPS) and reducing gut ammonia production (e.g. with lactulose, rifaximin) are both targets in management.
Causes of Portal Hypertension
Although cirrhosis is by far the most common cause, portal hypertension may arise at different anatomical levels. Classification by level helps localise the cause and guides investigation.
Portal hypertension can be classified by where resistance occurs in relation to the liver. This makes the cause easier to remember and helps distinguish conditions with preserved synthetic function (pre-hepatic) from those with hepatic parenchymal disease (intra-hepatic).
| Level | Meaning | Simple Memory |
|---|---|---|
| Pre-hepatic | Obstruction before blood enters the liver | Before liver |
| Intra-hepatic | Obstruction within the liver | Inside liver |
| Post-hepatic | Obstruction after blood leaves the liver | After liver |
Pre-hepatic
The obstruction occurs in the portal venous system before blood reaches the liver. Hepatic synthetic function is typically preserved.
- Portal vein thrombosis
- Splenic vein thrombosis
Intra-hepatic
The obstruction is within the liver parenchyma or sinusoids. Cirrhosis is the dominant cause worldwide.
- Cirrhosis (most common)
- Schistosomiasis (periportal fibrosis — most common cause globally in endemic regions)
- Nodular regenerative hyperplasia
Post-hepatic
The obstruction is in the hepatic veins or beyond, causing back-pressure into the portal system.
- Budd–Chiari syndrome (hepatic vein thrombosis)
- Right heart failure
- Constrictive pericarditis
| Level | Examples |
|---|---|
| Pre-hepatic | Portal vein thrombosis, splenic vein thrombosis |
| Intra-hepatic | Cirrhosis, schistosomiasis, nodular regenerative hyperplasia |
| Post-hepatic | Budd–Chiari syndrome, right heart failure, constrictive pericarditis |
Pre-hepatic causes preserve hepatic synthetic function — bilirubin, albumin and INR are often normal despite significant portal hypertension. This distinguishes pre-hepatic portal hypertension (e.g. portal vein thrombosis) from cirrhotic portal hypertension where synthetic function is impaired.
Common Misconceptions
False. Portal hypertension refers specifically to increased pressure in the portal venous system, not systemic arterial blood pressure. In fact, patients with portal hypertension often have systemic hypotension due to splanchnic vasodilation and reduced effective arterial volume.
False. Cirrhosis is the most common cause, but portal vein thrombosis, Budd–Chiari syndrome, schistosomiasis, and right heart failure can all cause portal hypertension — often with preserved hepatic synthetic function. The level of obstruction (pre-hepatic, intra-hepatic, post-hepatic) determines which other features are present.
False. Varices carry a bleeding risk, but not all varices bleed. Risk depends on variceal size, HVPG (bleeding risk rises above ≥12 mmHg), the presence of red wale signs on endoscopy, and the severity of the underlying liver disease. Small varices in well-compensated cirrhosis may be managed conservatively with surveillance.
False. Hypoalbuminaemia reduces oncotic pressure and contributes to ascites, but portal hypertension, splanchnic vasodilation, and renal sodium retention via RAAS and ADH activation are the central driving mechanisms. Treating only albumin without addressing portal pressure or sodium retention is insufficient.
Exam Tips
Portal vein = superior mesenteric vein + splenic vein
Normal HVPG = 1–5 mmHg
Portal hypertension = HVPG >5 mmHg
Clinically significant = HVPG ≥10 mmHg
Variceal bleeding risk = HVPG ≥12 mmHg
Most common cause = cirrhosis
Low platelets in cirrhosis = portal hypertension + hypersplenism
- Portal vein = SMV + splenic vein — know the anatomy.
- Two mechanisms in cirrhosis: structural (fibrosis/nodules — irreversible) and dynamic (reduced NO/vasoconstriction — modifiable).
- HVPG ≥10 mmHg = clinically significant; ≥12 mmHg = variceal bleeding risk.
- Collateral sites: gastro-oesophageal (varices), umbilicus (caput medusae), rectum (rectal varices).
- Low platelets in cirrhosis = hypersplenism + reduced thrombopoietin — not bone marrow failure alone.
- Ascites mechanism: portal hypertension → splanchnic vasodilation → RAAS/ADH → sodium/water retention.
- Pre-hepatic causes preserve synthetic function — bilirubin, albumin, INR often normal.
- Hepatic encephalopathy results from portosystemic shunting of gut-derived toxins bypassing the liver.
Frequently Asked Questions
One-Minute Portal Hypertension Revision
This summary brings together the most important pathway from cirrhosis to its clinical complications. Use it as a rapid revision tool before examinations.
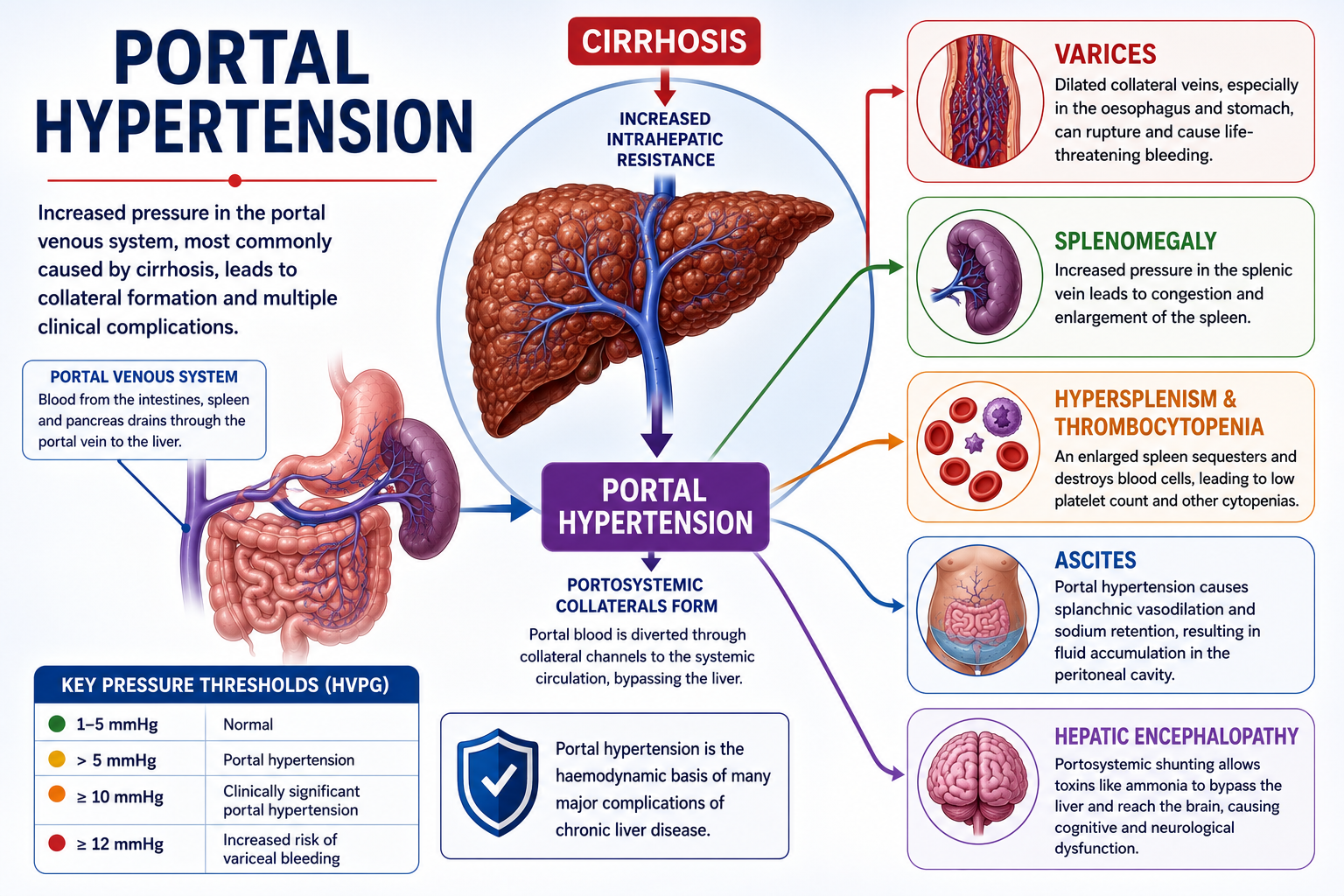
Key Takeaways
- Portal hypertension is increased pressure in the portal venous system
- Cirrhosis is the most common cause
- Increased intrahepatic resistance — structural and dynamic — is the key mechanism
- Portal hypertension causes portosystemic collateral formation
- Varices are dilated collateral veins with significant bleeding risk at HVPG ≥12 mmHg
- Splenomegaly occurs due to splenic venous congestion
- Thrombocytopenia commonly results from hypersplenism and reduced thrombopoietin
- Ascites results from portal hypertension, splanchnic vasodilation and sodium retention
- Portal hypertension is the foundation concept for understanding cirrhosis complications
References
- Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management. Hepatology. 2017;65(1):310–335.
- Bosch J, Abraldes JG, Fernández M, García-Pagán JC. Hepatic endothelial dysfunction and abnormal angiogenesis: new targets in the treatment of portal hypertension. J Hepatol. 2010;53(3):558–567.
- Ripoll C, Groszmann R, Garcia-Tsao G, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481–488.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–460.
- Runyon BA. Management of adult patients with ascites due to cirrhosis. Hepatology. 2009;49(6):2087–2107.
- D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231.
- de Franchis R, Bosch J, Garcia-Tsao G, Reiberger T, Ripoll C; Baveno VII Faculty. Baveno VII — renewing consensus in portal hypertension. J Hepatol. 2022;76(4):959–974.
This article is intended for medical education only. It is designed for medical students, intern doctors, and junior doctors and does not constitute clinical advice. Always refer to current local guidelines and specialist hepatological input when investigating and managing patients with portal hypertension.
- Portal Venous System
- What Is Portal Hypertension?
- Why Is It Dangerous?
- How Cirrhosis Causes PHT
- Portosystemic Collaterals
- Varices
- Splenomegaly & Hypersplenism
- Why Platelets Fall
- Ascites
- Clinical Consequences
- Hepatic Encephalopathy
- Causes
- Common Misconceptions
- Exam Tips
- FAQ
- One-Minute Revision
- Key Takeaways
- References