Most students memorise that hepatorenal syndrome is renal failure in cirrhosis. But the most important concept is this:
In HRS, the kidneys are usually structurally normal.
The problem is not primary kidney destruction. The problem is severe circulatory dysfunction caused by advanced liver disease. The kidneys fail because the circulation fails them — not because the kidneys themselves are diseased.
This article connects the pathophysiology you have already covered in Portal Hypertension Explained, Ascites Explained, and SBP Explained — and shows how they all converge in HRS.
HRS is functional renal failure. The kidneys fail because they are underperfused — not because they are structurally destroyed. This single insight explains the pathophysiology, the diagnosis, and the treatment of HRS.
Learning Objectives
- Define hepatorenal syndrome
- Explain why the kidneys are structurally normal in HRS
- Describe the haemodynamic cascade leading to HRS
- Distinguish HRS-AKI from HRS-NAKI (and old Type 1 / Type 2 terminology)
- Recognise common triggers, especially SBP
- Understand the diagnostic approach and exclusion criteria
- Differentiate HRS from ATN
- Explain why albumin and terlipressin are used
- Understand prognosis and the role of liver transplantation
What Is Hepatorenal Syndrome?
Hepatorenal Syndrome is functional renal failure occurring in advanced liver disease — usually cirrhosis with ascites — in the absence of another clear cause of kidney injury.
- The kidneys appear structurally normal on histology and imaging
- There is no major intrinsic kidney disease (no significant glomerulonephritis, interstitial nephritis, or tubular injury)
- Renal failure occurs because severe circulatory dysfunction causes intense renal vasoconstriction and reduced glomerular filtration rate (GFR)
failing because of a failing circulation
Why Is HRS Called Functional Renal Failure?
The term functional renal failure means that the kidneys are failing in function, but they are not primarily damaged in structure.
If a kidney from a patient with HRS were examined microscopically, major intrinsic renal disease would usually not be found. There is usually no significant glomerulonephritis, interstitial nephritis or tubular necrosis.
The problem is severe renal vasoconstriction caused by advanced liver disease and circulatory dysfunction. When circulation improves — for example after effective vasoconstrictor therapy or liver transplantation — renal function may recover.
Functional renal failure does not mean harmless or mild. HRS is life-threatening. "Functional" simply means the primary problem is haemodynamic and potentially reversible, rather than structural kidney destruction.
HRS is a diagnosis of exclusion. Before diagnosing HRS, other causes of AKI in cirrhosis must be excluded — volume depletion, nephrotoxins, obstruction, shock and intrinsic renal disease.
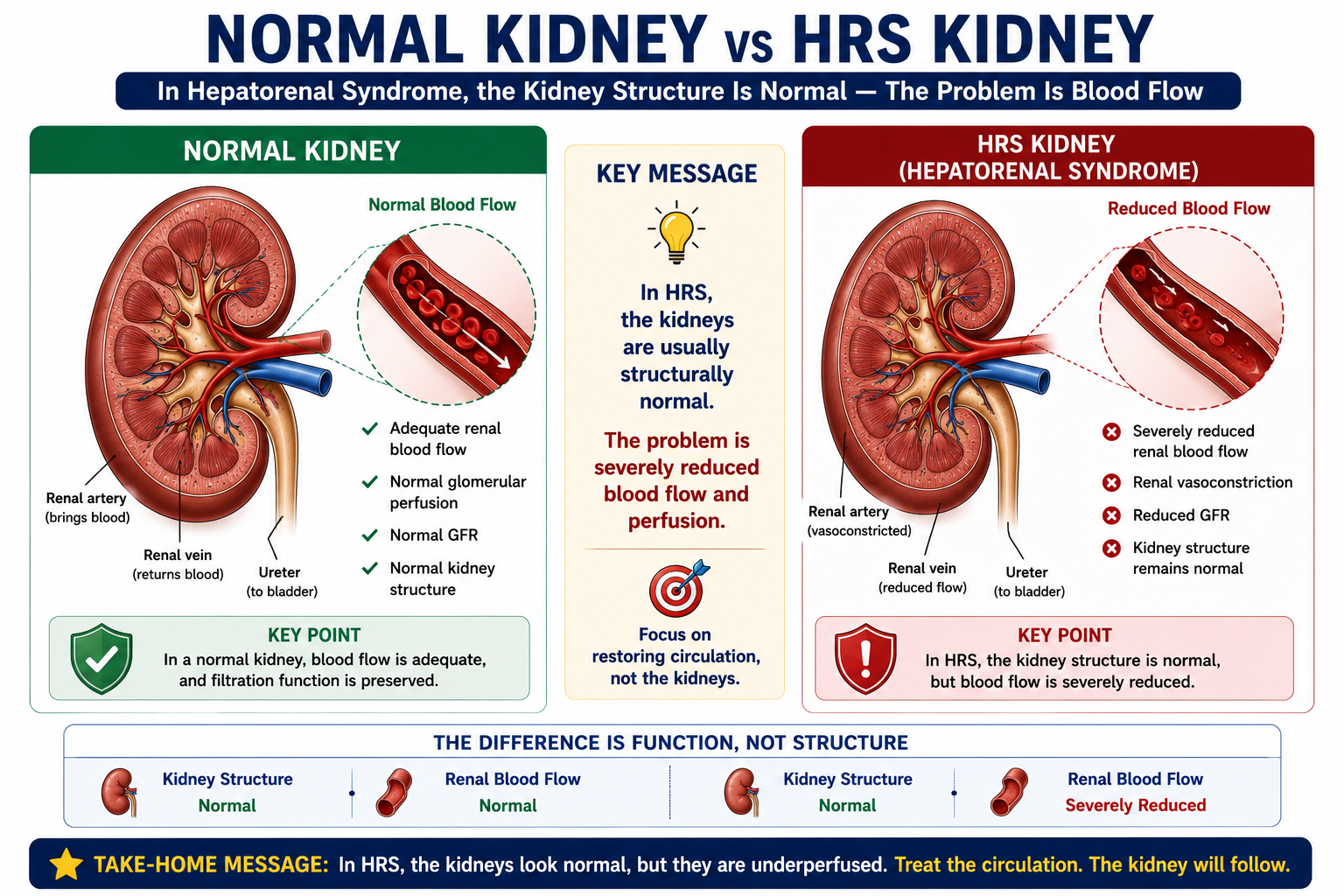
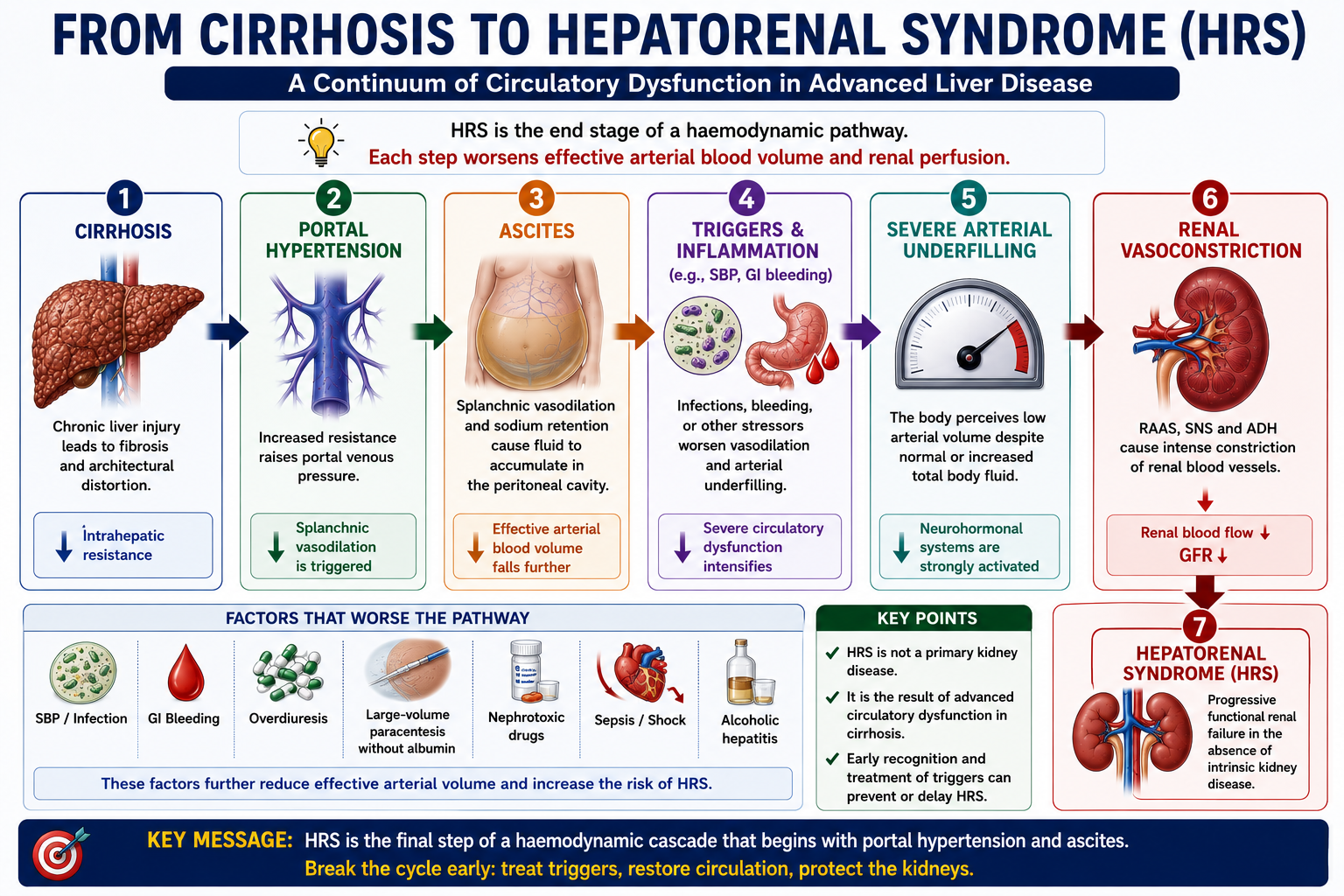
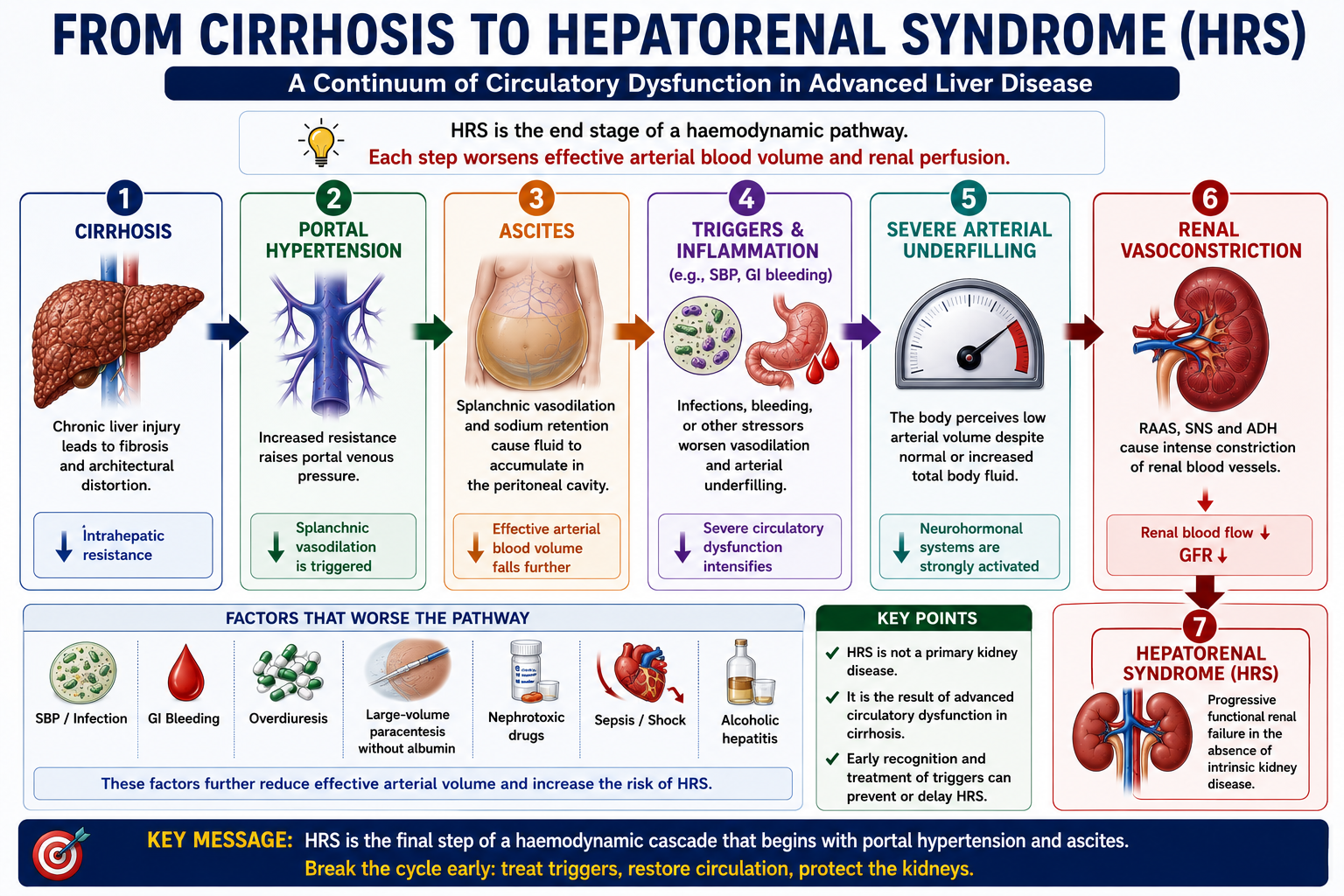
Why Does HRS Occur?
The pathophysiology of HRS is an extreme continuation of the same circulatory problem that produces ascites. As cirrhosis progresses, portal hypertension promotes increasing splanchnic vasodilation. Effective arterial blood volume falls progressively. When the body's compensatory systems can no longer maintain renal perfusion, HRS develops.
The kidneys are trying to preserve blood pressure and circulating volume, but this compensatory response becomes harmful. Intense renal vasoconstriction reduces renal plasma flow to the point where GFR falls critically.
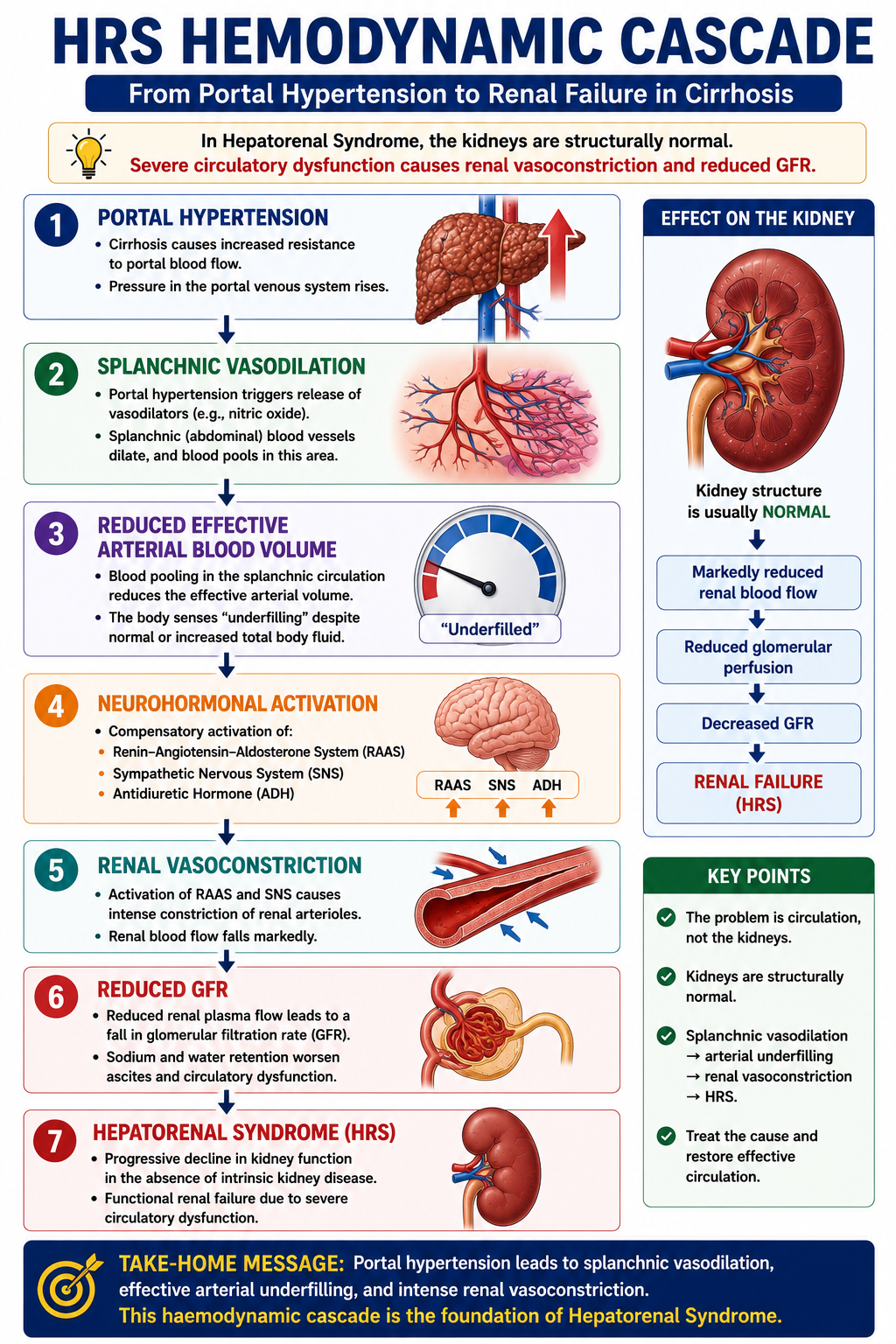
Pathophysiology of HRS
In cirrhosis, portal hypertension promotes release of vasodilators — especially nitric oxide — within the splanchnic circulation. Blood pools in the dilated splanchnic vascular bed. Although total body fluid may be increased (ascites, oedema), the effective arterial blood volume falls.
The body interprets this as underfilling.
In response, three major neurohormonal systems activate:
- Renin-angiotensin-aldosterone system (RAAS)
- Sympathetic nervous system
- Antidiuretic hormone (ADH/vasopressin)
Initially these responses help maintain arterial pressure and renal perfusion. In advanced disease they become excessive and maladaptive. Renal blood vessels constrict profoundly, renal plasma flow falls, and GFR declines — leading to HRS.
The kidney is not the primary problem. The kidney is responding to a failing circulation. This is why transplanting a liver — not a kidney — is the definitive treatment for HRS.
HRS-AKI and HRS-NAKI
Older teaching divided HRS into Type 1 (rapid onset) and Type 2 (more gradual). Modern terminology introduced by the International Club of Ascites uses:
| Older Term | Modern Term | Clinical Meaning |
|---|---|---|
| Type 1 HRS | HRS-AKI | Rapid rise in creatinine (doubling to >226 µmol/L within 2 weeks or rapid AKI criteria). High short-term mortality. |
| Type 2 HRS | HRS-NAKI | More gradual, sustained renal dysfunction that does not meet AKI criteria. Often associated with refractory ascites. |
Older textbooks and many examination questions still use Type 1 HRS and Type 2 HRS. Modern terminology uses HRS-AKI for acute presentations and HRS-NAKI for non-AKI presentations. In exams, recognise both systems and translate between them.
Many examinations still use the Type 1 / Type 2 terminology. Know both. Type 1 HRS = HRS-AKI (rapid, severe, high mortality). Type 2 HRS = HRS-NAKI (more gradual, often with refractory ascites).
Common Triggers of HRS
HRS often develops after a precipitating event that worsens circulatory dysfunction or renal perfusion. Recognising and treating the trigger is an essential part of management.
| Trigger | Mechanism |
|---|---|
| Spontaneous bacterial peritonitis (SBP) | Systemic inflammation worsens arterial underfilling and renal vasoconstriction |
| Sepsis / bacterial infection | Inflammatory mediators intensify splanchnic vasodilation and circulatory dysfunction |
| Gastrointestinal bleeding | Hypovolaemia reduces renal perfusion pressure acutely |
| Overdiuresis | Excessive fluid removal causes volume depletion and prerenal injury that can transition to HRS |
| Large-volume paracentesis without albumin | Post-paracentesis circulatory dysfunction (PPCD) worsens effective arterial volume |
| Nephrotoxic drugs (NSAIDs, aminoglycosides) | Direct renal vasoconstriction or tubular injury compound circulatory dysfunction |
| Severe alcoholic hepatitis | Acute-on-chronic liver failure causes profound circulatory and immune dysfunction |
Spontaneous bacterial peritonitis (SBP) is the most classically tested trigger of HRS. SBP causes systemic inflammation that worsens the circulatory dysfunction already present in cirrhosis, reducing effective arterial blood volume and precipitating renal vasoconstriction. This is why albumin is given alongside antibiotics in SBP — to reduce the risk of HRS. See SBP Explained.
Clinical Features
HRS does not usually produce specific kidney symptoms. The clinical picture is dominated by advanced cirrhosis with worsening renal function. Clinicians must think of HRS when a patient with known cirrhosis and ascites develops otherwise unexplained AKI.
- Cirrhosis with ascites (nearly always present)
- Rising serum creatinine
- Reduced urine output (oliguria)
- Low blood pressure or features of arterial underfilling
- Hyponatraemia — common, reflects water retention via ADH
- Peripheral oedema
- Hepatic encephalopathy — confusion or altered behaviour
- Recent trigger such as SBP, GI bleeding or sepsis
Think HRS when a patient with cirrhosis and ascites develops otherwise unexplained AKI. The absence of specific kidney symptoms and the presence of circulatory features should always prompt consideration of HRS as the underlying mechanism.
Diagnosis of HRS
HRS is a diagnosis of exclusion. Before HRS can be diagnosed, other causes of AKI in cirrhosis must be systematically excluded.
- Cirrhosis with ascites
- Acute kidney injury or worsening renal function
- No improvement after stopping diuretics and giving albumin
- No shock
- No current or recent nephrotoxic drug exposure
- No evidence of obstructive uropathy
- No significant proteinuria, haematuria or abnormal renal imaging suggesting intrinsic kidney disease
- 1Confirm cirrhosis and ascites — HRS almost always occurs in this context.
- 2Stop diuretics and nephrotoxic drugs — overdiuresis is a common reversible cause of AKI in cirrhosis.
- 3Perform diagnostic paracentesis — to exclude SBP as the precipitant (PMN ≥250 cells/mm³). See SBP Explained.
- 4Give albumin challenge — 1 g/kg/day for 2 days. If creatinine improves, the cause is likely volume depletion, not HRS.
- 5Exclude shock — HRS is not diagnosed in the presence of haemodynamic shock requiring vasopressors.
- 6Exclude obstruction — renal ultrasound should not show hydronephrosis or obstructive uropathy.
- 7Exclude intrinsic renal disease — urinalysis should show no significant proteinuria or haematuria. No features of glomerulonephritis or interstitial nephritis.
- 8If renal function does not improve despite above measures — HRS is the diagnosis.
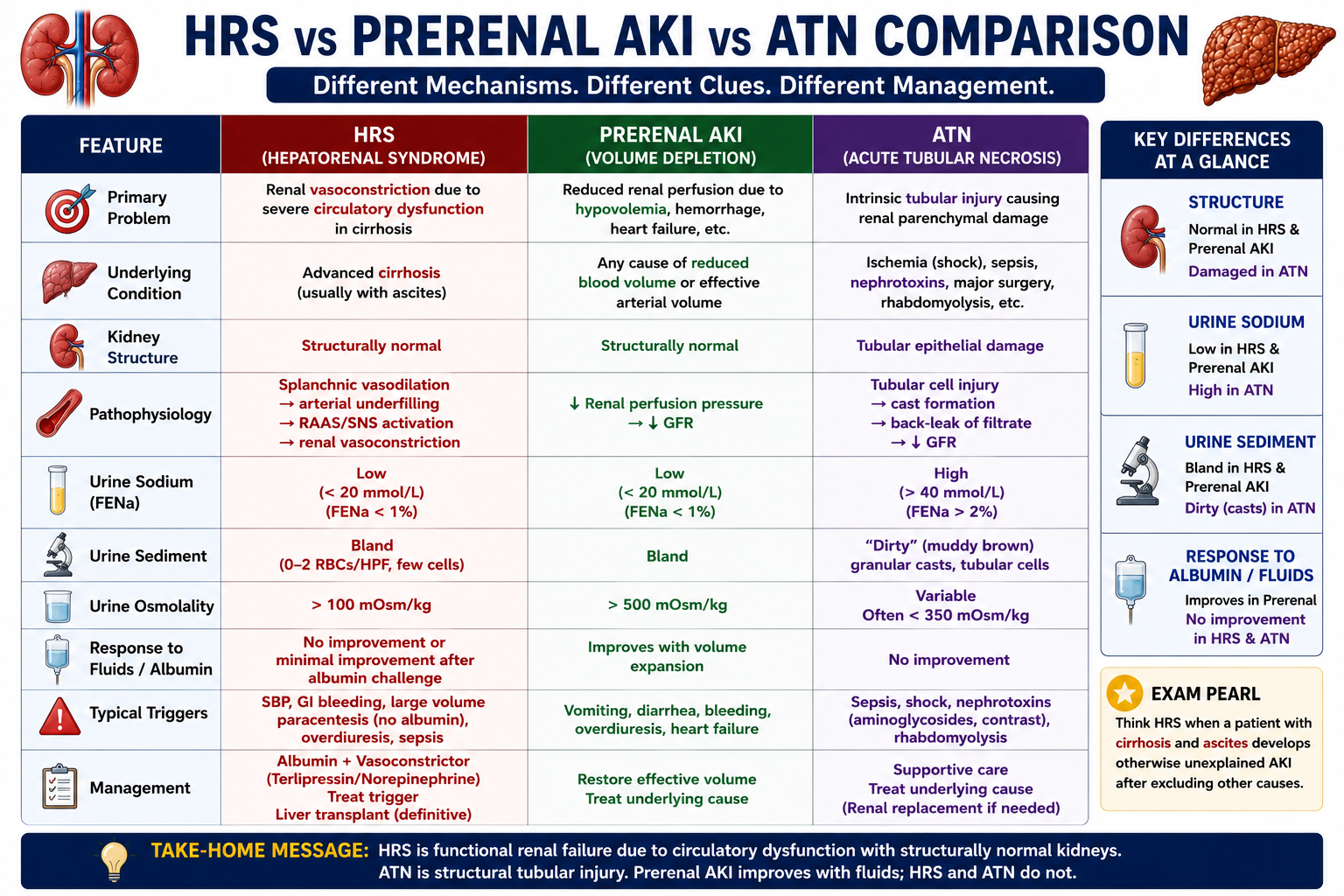
HRS vs ATN
Distinguishing HRS from acute tubular necrosis (ATN) is an important clinical and examination skill. Both cause AKI in cirrhosis but through fundamentally different mechanisms.
| Feature | HRS | ATN |
|---|---|---|
| Main problem | Renal vasoconstriction — functional | Tubular injury — structural |
| Kidney structure | Usually normal | Tubular cell damage and necrosis |
| Urine sodium | Usually low (<10 mmol/L) | Often high (>20 mmol/L) |
| Urine sediment | Bland — no casts | Granular (muddy-brown) casts |
| Fractional excretion of sodium | Low (<1%) | Often high (>2%) |
| Common trigger | SBP, sepsis, cirrhosis progression | Sepsis, shock, nephrotoxins |
| Treatment | Albumin + vasoconstrictor (terlipressin) | Supportive, treat underlying cause |
HRS = functional renal failure (vasoconstriction). ATN = structural tubular injury. Low urine sodium and bland urine sediment favour HRS. High urine sodium and granular casts favour ATN. Note: in advanced cirrhosis, interpretation of urine sodium can be unreliable — the full clinical picture is essential.
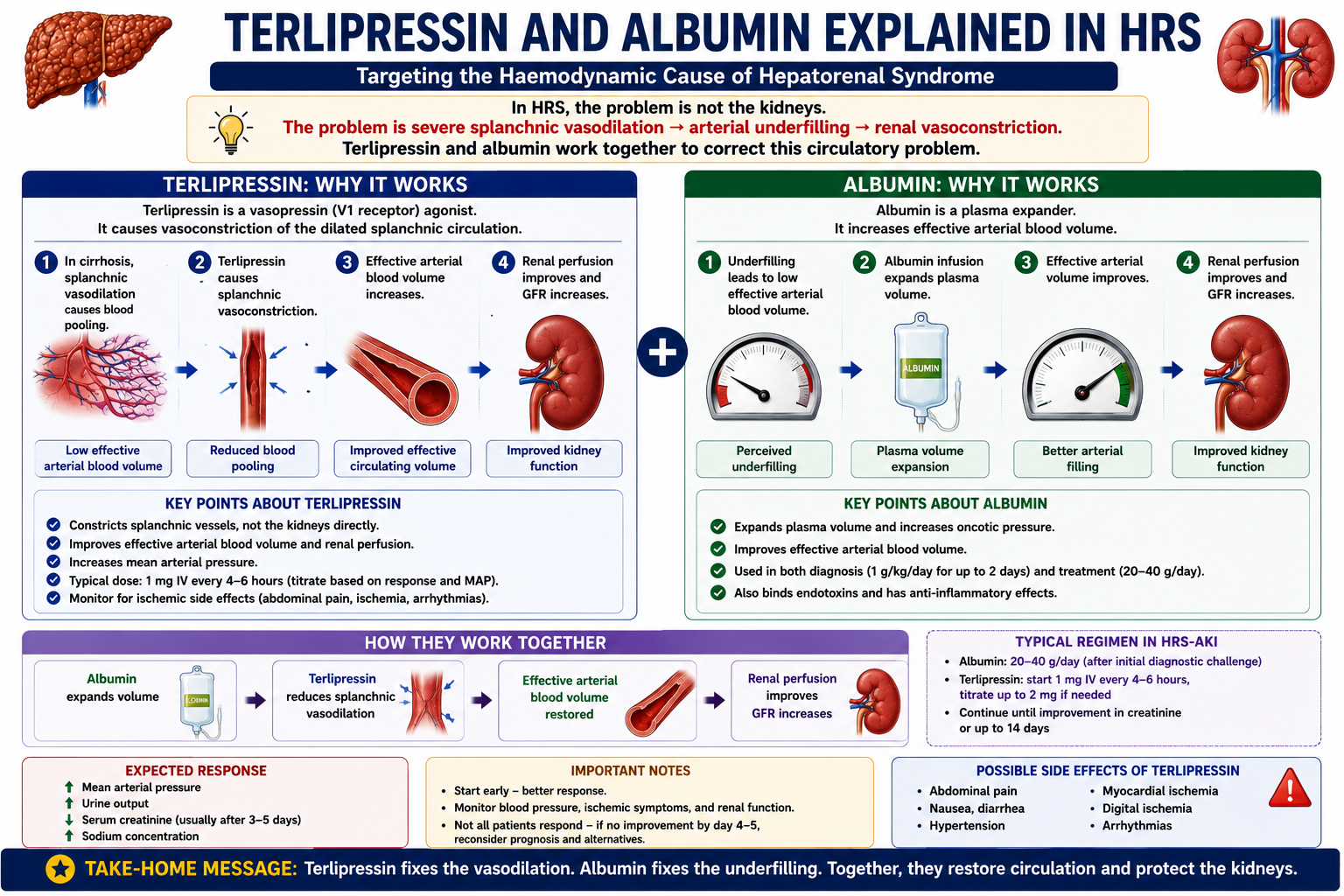
Why Albumin Is Used in HRS
Albumin serves two purposes in the management of HRS:
1. Diagnostic Use
As part of the diagnostic work-up, albumin is given as a volume challenge (1 g/kg/day for 2 days, capped at 100 g/day). If creatinine improves, the kidney injury was likely volume-responsive — prerenal AKI from over-diuresis or volume depletion — rather than true HRS.
2. Therapeutic Use
Albumin expands effective circulating volume and helps correct arterial underfilling. In HRS, this reduces the neurohormonal stimulus driving renal vasoconstriction and supports renal perfusion.
This is the same physiological principle as albumin use in SBP — improving effective arterial circulation to protect renal perfusion. In both conditions, the target is the failing circulation, not the kidney itself.
In SBP Explained, albumin is given to prevent HRS developing after SBP. Here in HRS, albumin is part of treatment once HRS has developed. The mechanism is identical — albumin expands effective arterial volume, reduces renal vasoconstriction, and protects GFR.
Why Terlipressin Works
Terlipressin is a vasopressin analogue that causes splanchnic vasoconstriction.
By constricting the dilated splanchnic vascular bed, terlipressin:
- Reduces blood pooling in splanchnic vessels
- Improves effective arterial blood volume
- Reduces the neurohormonal drive to renal vasoconstriction
- Improves renal perfusion and GFR
Albumin expands volume. Terlipressin corrects pathological vasodilation. Together they target the haemodynamic cause of HRS from two complementary angles — both essential because neither alone is sufficient in most cases.
Prognosis and Definitive Treatment
HRS has a poor prognosis, especially HRS-AKI, unless the underlying liver disease is treated. Medical therapy with albumin and terlipressin may improve renal function temporarily — buying time — but does not address the underlying cause.
HRS indicates advanced decompensated cirrhosis. The development of HRS is associated with a median survival of weeks to months without definitive treatment.
Liver transplantation is the definitive treatment for HRS in suitable patients. Transplanting the liver restores normal hepatic function, resolves portal hypertension, reverses splanchnic vasodilation and neurohormonal activation, and allows the structurally normal kidneys to recover function. This confirms the core principle: HRS is a hepatic and circulatory problem, not a renal one.

One-Minute HRS Revision
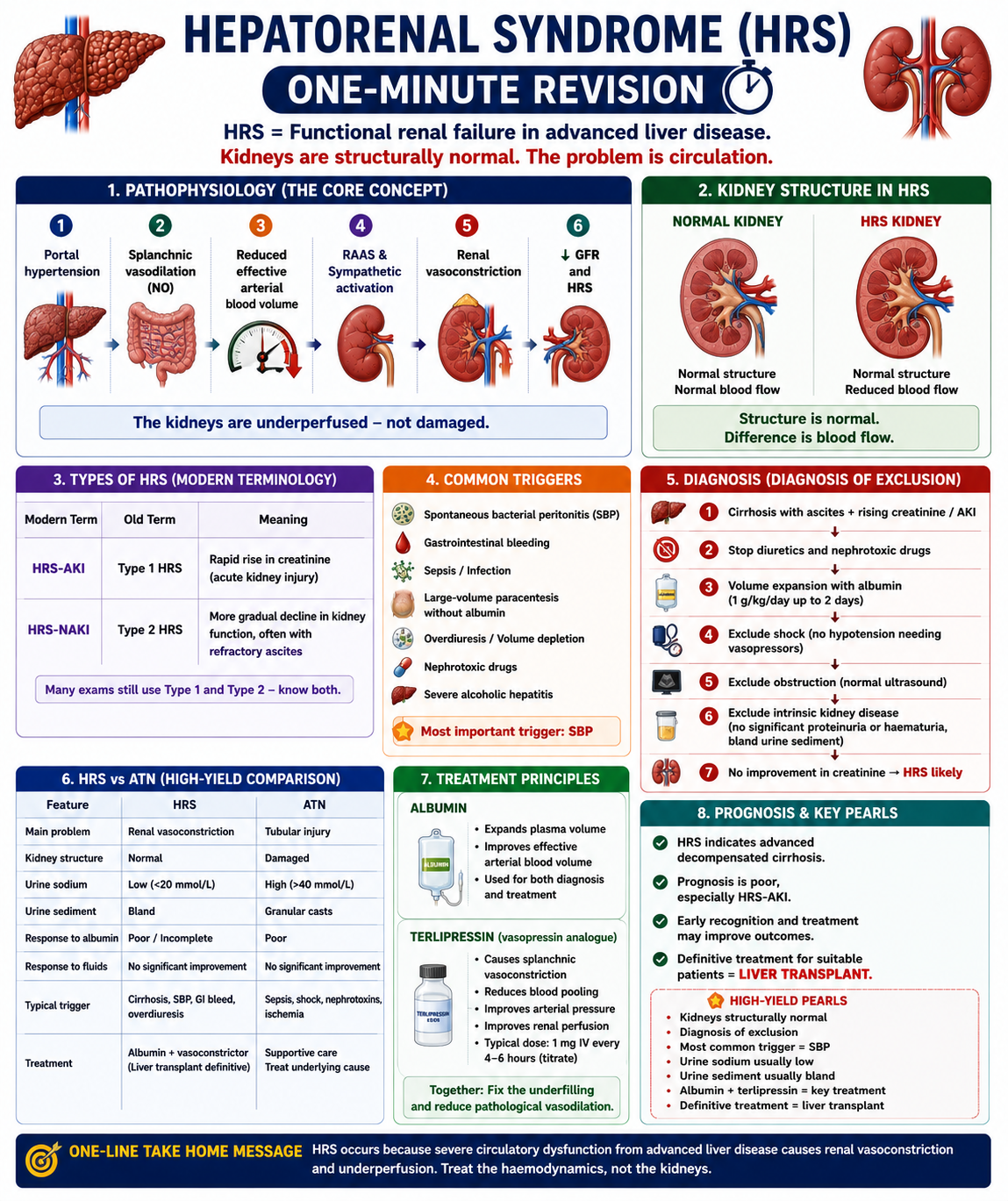
High-Yield Exam Pearls
HRS = functional renal failure in cirrhosis
Kidneys are structurally normal
Diagnosis of exclusion
Most important trigger = SBP
Urine sodium usually low (<10 mmol/L)
Urine sediment usually bland
HRS-AKI = Type 1 (rapid, severe)
HRS-NAKI = Type 2 (gradual, refractory ascites)
Treatment = albumin + terlipressin
Definitive treatment = liver transplantation
- HRS is functional, not structural — the kidneys are normal; the circulation is the problem. This explains why liver transplant cures it.
- Diagnosis of exclusion — always exclude volume depletion, nephrotoxins, obstruction, shock and intrinsic renal disease first.
- Albumin challenge — if creatinine improves with albumin, the diagnosis is more likely volume-responsive prerenal AKI than HRS.
- SBP is the most important trigger — always tested. This is why albumin is given in SBP even before HRS develops.
- Low urine sodium favours HRS over ATN, but is not absolute — use the whole clinical picture.
- Terlipressin targets splanchnic vasodilation — not the kidney directly. The mechanism is haemodynamic correction.
- Functional does not mean mild — HRS is functional because the kidney structure is preserved, but it is still life-threatening.
- Ascites is central to diagnosis — classic HRS occurs in cirrhosis with ascites; renal failure without ascites should prompt careful reconsideration.
- Transplant the liver, not the kidney — because the kidney is underperfused rather than structurally destroyed.
- Liver transplant is the only cure — always state this when asked about definitive management.
- NSAIDs are contraindicated in cirrhosis — they block prostaglandin-mediated renal vasodilation, precipitating or worsening HRS.
Key Takeaways
- HRS is functional renal failure in cirrhosis — the kidneys are structurally normal
- Functional renal failure means the kidneys are failing because of haemodynamic dysfunction, not primary structural destruction
- Modern diagnostic criteria require cirrhosis with ascites, AKI/worsening renal function, no response to albumin, and exclusion of shock, nephrotoxins, obstruction and intrinsic kidney disease
- Liver transplantation is definitive because it corrects the liver-driven circulatory abnormality causing renal vasoconstriction
- The fundamental mechanism is splanchnic vasodilation causing reduced effective arterial blood volume and renal vasoconstriction
- RAAS, sympathetic nervous system, and ADH activation drive renal vasoconstriction
- HRS-AKI (Type 1) is rapid and severe; HRS-NAKI (Type 2) is more gradual
- SBP is the most important and classically tested trigger of HRS
- HRS is a diagnosis of exclusion — volume depletion, nephrotoxins, obstruction and intrinsic renal disease must be excluded first
- Albumin serves both diagnostic (volume challenge) and therapeutic roles
- Terlipressin works by constricting the splanchnic vasculature, improving effective arterial volume and renal perfusion
- Low urine sodium and bland urine sediment favour HRS over ATN
- NSAIDs are contraindicated in cirrhosis — they precipitate HRS
- HRS indicates advanced decompensated cirrhosis with poor prognosis without transplant
- Liver transplantation is the definitive treatment for HRS
Frequently Asked Questions
References
- Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361(13):1279–1290.
- Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56(9):1310–1318.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–460.
- Runyon BA; AASLD. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57(4):1651–1653.
- Angeli P, Ginès P, Wong F, et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J Hepatol. 2015;62(4):968–974.
- Sort P, Naveau M, Arroyo V, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341(6):403–409.
- Moreau R, Lebrec D. The use of vasoconstrictors in patients with cirrhosis: type 1 HRS and beyond. Hepatology. 2006;43(3):385–394.
This article is intended for medical education only. It is designed for medical students, intern doctors, and junior doctors and does not constitute clinical advice. Always refer to current local guidelines and specialist hepatological input when managing patients with suspected HRS.